The hidden treasure in RNA-seq
Michael Stadler and his team at the Friedrich Miescher institute for Biomedical Research (FMI) have developed a novel computational approach to analyze RNA-seq data. By comparing intronic and exonic RNA reads, this approach allows discerning the contribution of transcriptional and post-transcriptional regulation to gene expression. The description of this new method and the results of the analysis have been published in Nature Biotechnology.
RNA-seq offers an important snapshot of cell state. It tells scientists what genes are active and how active they are. If the scientists then compare the RNA-seq snapshots of for example diseased and healthy cells, young and old cells, or mature and stem cells, they can start to hypothesize about the genes involved in the disease, in aging or in cell differentiation.
Various processes control the accumulation of RNA in a cell throughout its lifecycle. Some processes directly control RNA production (transcriptional), others take place as the RNA is processed, matures and is finally degraded (post-transcriptional). All of them have an influence on the fate of the cell, but RNA-seq is usually applied to measure RNA levels without distinction between these processes.
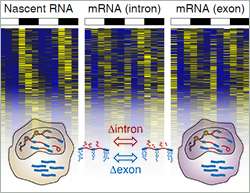
Dimos Gaidatzis, Lukas Burger and Michael Stadler have now developed a computational approach called EISA (exon-intron split analysis) that measures the change in mRNA and pre-RNA levels across different conditions. By doing so the computational biologists could distinguish between RNAs that are regulated at the transcriptional level鈥攁s it is being transcribed from the genome, or at the post-transcriptional level鈥攁s it matures or is being degraded.
"In one of our projects for a group here at the FMI we observed that reads arising from exons lagged behind the reads from introns," explained Gaidatzis, "it looked as if the intronic reads are a more direct measure of transcription. Thus we wanted to test if this is true in general." The scientists analyzed 17 published RNA-seq datasets of different cell types and biological situations. They showed that most intronic RNA reads arise from RNA molecules that are still in the nucleus and have just been transcribed. Changes in these reads accurately account for changes in transcriptional activity. In addition, a comparison between intronic and exonic reads, allows predictions about post-transcriptional regulation.
"EISA allows us to dissect the contribution of transcriptional and post-transcriptional processes on changes in gene expression," said Stadler. "With this, the amount of information gained from a single RNA-seq data set has increased, we have indeed found a hidden treasure in RNA-seq."
More information: "Analysis of intronic and exonic reads in RNA-seq data characterizes transcriptional and post-transcriptional regulation." Nature Biotech.
Journal information: Nature Biotechnology

















