September 23, 2019 feature
Unfolding adsorption on metal nanoparticles: Connecting stability with catalysis

Thamarasee Jeewandara
contributing writer
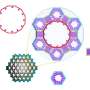
 calculations performed. Upper: Coordination numbers on (A) 55-atom icosahedron, (B) 55-atom cuboctahedron, (C) 147-atom icosahedron, (D) 147-atom cuboctahedron, (E) 172-atom cube. Nanoparticles (NPs) where more than one unique atom share the same coordination number (CN), are denoted with numbers 8-1, 8-2, 8-3, 8-4. Credit: Science Advances, doi: 10.1126/sciadv.aax5101")
Metal nanoparticles have received substantial attention due to their applications in diverse fields from medicine, catalysis, energy and the environment. However, the fundamental properties of nanoparticle adsorption on a surface remain to be understood. James Dean and an interdisciplinary research team in the department of Chemical Engineering, in the U.S. introduced a universal adsorption model to account for the structural characteristics, metal composition and different adsorbates of nanoparticles via machine learning (ML). The model fit a large number of data to accurately predict adsorption trends on monometallic and alloy-based nanoparticles. The template was simple and provided rapidly calculated data for metals and adsorbates. The research team connected the adsorption with stability behavior to advance the design of optimal nanoparticles for applications of interest. The research is now published on Science Advances.
Metal nanoparticles (NPs) have significant applications in catalysis, ranging from , to . But their stability and catalytic activity generally show opposing trends, where very active catalysts can only operate for a few cycles. A key feature on the extent of metallic catalytic functionality depends on the strength of adsorption for a variety of species on the catalyst surface. According to the , developed more than a century ago, active catalysts should bind adsorbates with a binding strength that is neither strong nor weak. While strongly adsorbed species can poison the catalyst surface, weakly bound reactants desorb easily. In an intermediate scenario, the reactants can meet each other and react on the catalytic surfaces. Researchers currently use and theoretical chemistry methods to stimulate catalytic behavior on metal catalysts with great accuracy to guide subsequent experiments in the lab.
Computational efforts have focused on screening different metal catalysts to discover the "magic" binding energy (BE) of chemical species on catalyst surfaces to form very active catalysts. The in silico design of catalytically active materials, however, remains to be realized. The drawbacks are mainly due to design efforts that often neglect the stability of catalysts. NP catalysts also possess a high degree of site heterogeneity on their surface for adsorption and catalysis. Scientists had developed adsorption models to relate binding energy of the adsorbates with surface characteristics of NPs such as (CNs) to understand the site-specific adsorption response. Yet, for clarity, the binding energy (BE) variation also entails secondary descriptors such as and of NPs.
 as a descriptor for adsorption energy. (A) The BE of CO on various sites of Au NPs as a function of CElocal: 172-atom cube (rectangles), 147-atom icosahedron (hexagons), and 147-atom cuboctahedron (rhombus). Heat map of different sites on the NPs with respect to their BE of CO (B to D) and to their CElocal (E to G). The color scheme follows the range of strongest CO binding to weakest CElocal (violet) and of weakest binding to strongest CElocal (red). Credit: Science Advances, doi: 10.1126/sciadv.aax5101")
In the present work, Dean et al. applied (DFT) and machine learning techniques to derive a simple physics-based model to accurately capture the varying adsorption energy. They estimated the variable as a function of the local adsorption site environment on the NP surface as well as the type of metal NP. The generalized model could be applied to any metal nanostructure to understand adsorption behavior on the NP catalyst and the stability of the catalyst; to screen and .
The researchers first hypothesized the most important factors between monometallic NPs and adsorbates. They then defined the (CElocal) in bulk metals and captured CEs in NPs using a , which summed every metal-metal bond energy. By applying similar concepts, they described the stability of binding sites and showed how chemically unsaturated sites (fewer metal-metal bonds) bound adsorbates with an increased strength. The research team focused on describing the binding capacity of a single adsorbate-metal pair. They plotted the DFT-calculated binding energy of carbon monoxide (CO) to a 172-atom gold (Au) cube and a 147-atom gold (Au) cuboctahedron or icosahedron. The team observed a strongly inverse relationship between the local cohesive energy (CElocal) and binding energy (BE) to suggest the strongest adsorption sites to be those exhibiting the weakest local cohesion.
The team further developed their model and performed (OLS) regression to understand adsorption on monometallic NPs and slabs using three adsorbates [Methyl radical (CH3), CO, hydroxyl radical (OH)] on three different metals (Cu, Ag—silver, Au). The metallic NPs contained different morphologies (172-atom cube, 55- and 147-atom icosahedron and 55- and 147-atom cuboctahedron). They observed that the binding affinity to the adsorbates decreased as the cohesion of the local sites increased. And as the adsorbate's chemical potential increased, they became less stable and bound a metal NP with higher tendency. The direct correlation with the metal Adsorbate (MAD) intuitively described the tendency of the metal to bind the adsorbate.
 of adsorbates (OH, CO, and CH3) on various metal systems versus the DFT BE (eV). (A) The model both trained and tested on PBE DFT data for NPs (Au/Ag/Cu, 55 to 172 atoms), which includes the nanoparticle cohesive energy (CENP) term. (B) The model both trained and tested on PBE DFT data for NPs (Au/Ag/Cu, 55 to 172 atoms), which does not include the CENP term. (C) The model trained on PBE DFT data for NPs (Au/Ag/Cu, 55 to 172 atoms) and tested against RPBE (revised Perdew-Burke-Ernzerhof model) DFT data for top-site adsorptions on metal surfaces (Au/Ag/Cu). (D) The model both trained and tested on RPBE DFT data for top-site adsorptions on metal surfaces (Au/Ag/Cu) from the slab dataset. Credit: Science Advances, doi: 10.1126/sciadv.aax5101")
Dean et al. tested the generalizability of the model and trained the simulation on a single metal or single morphology, although it accurately captured other metals or morphologies as well. The work provided strong evidence that the model captured the underlying physics of the binding interactions, allowing the team to extend the work from non-periodic NPs to periodic slab systems. Computationally inexpensive systems could parameterize the model to extend to larger systems, which was not thus far possible due to the computational costs involved.
Dean et al. then extended the model from monometallic NPs to bimetallic systems. For these experiments, they plotted the BE—trained on monometallic NPs, across several sites of bi-metallic, 55-atom icosahedron NPs (Cu31Ag24 and Cu22Ag33). The model very accurately captured trends in adsorption on the bimetallic Cu/Ag NPs as well. This was an interesting result since the scientists had only trained the model on monometallic systems. The results showed the generalizability of the model for both monometallic and bimetallic NPs. However, the team will account additional descriptors including binding site electronegativity to understand the adsorption behavior for bimetallic systems in depth.
 NPs. The model is trained on CH3, CO, and OH adsorbing on monometallic Ag, Cu, and Au NPs and is able to capture adsorption on bimetallic NPs. Images of the two NPs are shown as inset, with copper and silver atoms colored in brown and gray, respectively. Credit: Science Advances, doi: 10.1126/sciadv.aax5101")
Although Dean et al. trained the ML (machine learning) algorithm to capture the adsorption trends of just one type of , it could accurately predict the behavior of similar d9 metals (Cu—coper, Ag and Au). When they trained the model on a dataset of CH3, CO and OH adsorbed to Cu, Ag and Au NPs, they could also capture general adsorption trends for similar elements in other columns of the periodic table. They then improved the complexity of the machine learning techniques to provide additional avenues to improve the model of adsorption.
In this way, James Dean and his colleagues developed a simple yet powerful physics-based model to capture trends on the strength of binding interactions between different adsorbates and metal NPs using machine learning techniques. The study was the first to develop an adsorption model that accurately connected the properties of diverse metal NPs with the stability of the adsorption site. The model introduced simple descriptors to capture the adsorption on any site, relative to monometallic and bimetallic NPs. The team generalized the model to effectively stimulate a range of binding interactions, including variations on the types of metals, their composition, sites of adsorption and adsorbates.
 The model trained on the slab dataset on Cu, Ag, and Au surfaces and tested against the Rh, Ir, Ni, Pd, Pt, Cu, Ag, and Au surfaces from the slab dataset. (B) The equivalent model when trained separately for each column of the d-block, still using the slab dataset. Error bars in every case are the 10-fold cross-validated RMSE of the training set. RIGHT: Extension of the model to Rh and NH3. (A) The model parameterized on our Ag, Cu, and Au NPs adsorbing CH3, CO, and OH and tested against Rh and NH3. (B) The equivalent model with empirical (constant) corrections for Rh and NH3. In the case of NH3 bound to Rh, both corrections are simultaneously applied and indicated by two-colored dots. (C) The model trained on CH3, CO, OH, and NH3 adsorbing on icosahedral/cuboctahedral Rh55. Credit: Science Advances, doi: 10.1126/sciadv.aax5101")
Although the team did not test the applicability of the model for ternary systems, the physical properties may remain relevant to accurately model multimetallic systems as well. The adsorption model can accurately describe the binding strength of a variety of molecules on any site of NPs, including alloys. The scientists expect the model to be highly applicable as a screening tool for the high throughput search of potential catalysts.
Written for you by our author —this article is the result of careful human work. We rely on readers like you to keep independent science journalism alive. If this reporting matters to you, please consider a (especially monthly). You'll get an ad-free account as a thank-you.
More information: James Dean et al. Unfolding adsorption on metal nanoparticles: Connecting stability with catalysis, Science Advances (2019).
M. Valden. Onset of Catalytic Activity of Gold Clusters on Titania with the Appearance of Nonmetallic Properties, Science (2002).
Jeff Greeley et al. Alloy catalysts designed from first principles, Nature Materials (2004).
Journal information: Science Advances , Nature Materials , Science
© 2019 Science X Network