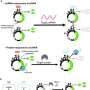
TEMPOmap reveals subcellular RNA kinetic landscape across RNA lifespan and cell cycle. a, Dynamic model for estimating RNA kinetic parameters. For each gene, RNA synthesis (α) and degradation constant (β) were estimated using single-cell RNA concentration. The export constant (λ) was estimated using the subcellular RNA reads. b, The dynamic model for estimating cytoplasmic translocation (γ) using DR-based analysis (Methods). c, Upper right, mathematical model of RNA life cycle and kinetic assumptions used for the parameter estimation. Bottom left, histogram of the four parameters for all genes that passed qualify control and scatterplots depicting the pairwise correlation of parameters with r value (Pearson correlation) and linear fitting curve. Color intensity of the dots indicates local density. Units of α is RNA concentration per hour, where RNA concentration is defined by copies per voxel (voxel = 200 nm × 200 nm × 350 nm). Units of β, λ, γ are per hour. d, Heatmap depicting pairwise correlation matrix of the four parameters estimated using single cells from three cell-cycle phases (G1, G1/S, G2/M). Color indicates the value of Pearson correlation coefficients. Boxed regions indicate the correlations of each parameter among three cell-cycle phases. Credit: Nature Methods (2023). DOI: 10.1038/s41592-023-01829-8
RNA plays a central role in biology, but there is still much to learn about the molecule's life cycle in the cell. In recent years, scientists have devised methods that can take snapshots of messenger RNAs (mRNAs) scattered across a cell and measure the ebb and flow of their abundance, but they haven't yet been able to track the individual movements of a large number of them over time.
A new method built by scientists at the Broad Institute of MIT and Harvard is the first large-scale approach to track mRNAs over both space and time in individual cells. Known as TEMPOmap (temporally resolved in situ sequencing and mapping), the method simultaneously measures the subcellular movements of many RNA molecules, with the potential to follow a molecule from its birth through to its death.
TEMPOmap chemically labels newly made RNA molecules and uses a unique approach to sequence them at specific locations in the cell at several time points, allowing researchers to track thousands of RNA molecules in parallel over time. In a study in Nature Methods, the research team described TEMPOmap and how it enabled them to follow mRNAs in a variety of cell types, measure the speed at which the molecules were transcribed from DNA and move within the cell, and discover surprising movements of the RNAs in the cells.
The scientists said maps generated by the approach can help uncover new principles of gene regulation and explore how RNA life cycle patterns influence the function of various cell types in health and disease.
"New ways of measuring the patterns of RNA movement and stability can provide fresh insights into basic principles of molecular biology, in addition to helping us better understand how RNA kinetics impact the function of cells across many different human cell types," said senior study author Xiao Wang, who is a core institute member and Merkin Institute Fellow at the Broad and an assistant professor of chemistry at MIT.
Time will tell
TEMPOmap builds upon STARmap, a method previously developed by Wang and colleagues that profiles the location of RNA molecules within a cell at one point in time. With TEMPOmap, the Wang lab devised an approach that tags only newly made RNAs as they are being transcribed from DNA, anchors them in place by fixing the cells in a hydrogel, and detects the RNAs at those locations in the cell. By doing so at different time points, the researchers infer the movement of RNA molecules through the cell over several hours.
To detect and sequence the RNAs in individual cells, the team first designed an inventive three-part chemical probe set that precisely binds to the label on a newly made mRNA, forming a circular structure that generates a tight ball of DNA fragments, also known as a "DNA amplicon," copied from the bound mRNA. The scientists then use an in situ sequencing method known as "SEDAL," which was devised for the STARmap approach, to analyze the DNA fragments and uncover the identity of the mRNA that's present at each spot.
The researchers demonstrated the method's potential by following the subcellular movements of mRNAs representing nearly 1,000 genes in cells in the lab. They also followed mRNAs for several dozen genes in fibroblasts and cardiomyocytes created from reprogrammed human stem cells, and more than 250 genes in human skin cells.
The analysis revealed that mRNAs for genes with a specific function in a specific cell type tend to be synthesized faster and remain more stable in that cell type than in others. "With TEMPOmap, we're able to see evidence for these transcripts having a highly functional role in particular cell types," said co-first author Jingyi (Rena) Ren, a graduate student at MIT and in the Wang lab.
The team's measurements also revealed the speed of each RNA's transcription, export from the nucleus, movement through the cytoplasm, and decay. They found that mRNAs displayed different kinetic patterns related to the molecular functions of the genes they encode.
In addition to uncovering new insights into gene expression and control, TEMPOmap can assist studies of other biological phenomena, such as embryonic development, biological clocks, and disease progression. The Wang lab aims to use spatial transcriptomic tools to better understand brain function and dysfunction at the molecular level. For example, they plan to use TEMPOmap to study how gene transcripts travel within brain cells under certain conditions, for example, after a neuron fires.
"Biology is, by nature, a dynamic process," said Ren. "By following these RNAs, we can help answer some fascinating biological questions."
More information: Jingyi Ren et al, Spatiotemporally resolved transcriptomics reveals the subcellular RNA kinetic landscape, Nature Methods (2023).
Journal information: Nature Methods
Provided by Broad Institute of MIT and Harvard