New method tracks gene expression changes to reveal cell fate decisions

Andrew Zinin
lead editor

Essentially all cells in an organism's body have the same genetic blueprint, or genome, but the set of genes that are actively expressed at any given time in a cell determines what type of cell it will be and its function. How rapidly gene expression in a single cell changes over time can provide insight into how cells might become more specialized, but current measurement approaches are limited. A new method developed by researchers at Penn State and Yale University incorporates spatial information from the cell as well as data from cells processed at different times, improving researchers' ability to understand the nuances of gene expression changes.
A paper describing the method, called spVelo, is published in the journal It calculates RNA velocity, which describes the direction and rate of change during transcription—a step of gene expression that involves copying the genetic code.
"Different sets of genes are expressed in a cell when they are activated, when they respond to stimuli and during the process of differentiation, which allows cells to develop into specific cell types," said Wenxin Long, a doctoral student in statistics in the Penn State Eberly College of Science and an author of the paper. "RNA velocity has emerged as a way to measure the how the rate of gene expression changes in a cell, which can tell us important information about the cell's current state and its future. Our new method overcomes important challenges of previous methods, making it a promising and robust way to calculate RNA velocity and learn more about the many functions of a cell."
During gene expression, DNA is first transcribed into messenger RNA (mRNA), which carries the genetic code that will be used to make proteins. But not all of the mRNA sequence is used; it must first undergo a process called splicing, which removes segments called introns that don't carry coding information, and splices back together the exons that do. The spliced mRNAs can then be translated into a protein sequence.
Using a technique called single-cell RNA sequencing (scRNA-seq), researchers can count the number of RNA strands that are spliced and those that are not yet spliced. By modeling the relationships between spliced and unspliced RNA abundance, researchers infer whether a gene is being upregulated and downregulated. The researchers said that this rate of spliced expression change—RNA velocity—is essentially a snapshot of the genes that are actively being turned on or off in the cell and can be used to infer future gene expression.
"A researcher can sequence the RNA from many cells at the same time, but cells processed at a later date or by different people or research groups can experience slightly different lab conditions that might impact the results," Long said. "It has been a challenge to incorporate multiple batches in one analysis. Our method can account for differences across multiple batches, so we can integrate a much larger amount of data in one analysis."
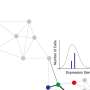
In addition to processing multiple batches at once, spVelo incorporates important spatial information from the cell, Long said.
"Newer types of sequencing data can provide spatial information, such as where the cell is located within a tissue," said Lingzhou Xue, professor of statistics in the Penn State Eberly College of Science and a co-corresponding author of the paper. "Some previous methods to calculate RNA velocity have been able to incorporate either spatial information or multiple batches, but not both. Combining the two allows us to glean the most information from large-scale, multi-batch spatial datasets."
The new method takes advantage of two types of neural networks—a type of machine learning—to overcome previous limitations. One of these neural networks, called a Variational Autoencoder, models gene expression. The second neural network, called a Graph Attention Network, allows the researchers to incorporate spatial and batch information from the sequencing data. The model also accounts for differences between batches using what is called a maximum mean discrepancy penalty, which enables RNA velocity inference across multiple datasets.
The researchers benchmarked spVelo with a variety of previous methods using a dataset of gene expression from oral squamous cell carcinoma, a type of cancer, as well as a simulated spatial dataset of pancreas cells that is commonly used by researchers to test and compare methods. The researchers said spVelo performed as well as or better on a variety of parameters. The method, they said, was also able to provide more complex trajectory patterns for a cell, suggesting future expression patterns and possible cell types or subtypes that a cell might differentiate into.
"Another advantage of our method is that it gives us a measure of confidence around our predictions, which previous methods lacked," Long said. "For example, we are pretty confident that some cells will remain as or transition to a particular cell type or subtype, while others might have more possibilities for transitioning."
The researchers said that the method could also be used to explore gene regulatory networks. For example, to understand the impact of a particular gene on a cell's fate, researchers could compare RNA velocities in a normal cell and in a cell where that gene has been deleted. Additionally, because RNA velocity provides information at a specific point in time, changes in RNA velocity over time could lend insight into how cells communicate with each other and at what rates.
"RNA velocity is still an emerging concept, and we believe there are a wide variety of applications," Xue said. "Having this more robust and reliable way to measure multiple batches and incorporate spatial data opens up new opportunities, and we are excited to see how our method is used in the future."
In addition to Long and Xue, the research team includes Tianyu Liu and Hongyu Zhao at Yale University. Funding from the National Institutes of Health supported this research.
More information: Wenxin Long et al, spVelo: RNA velocity inference for multi-batch spatial transcriptomics data, Genome Biology (2025).
Journal information: Genome Biology
Provided by Pennsylvania State University