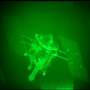
Graphic: Christine Daniloff
All living tissue is made from proteins, and all proteins are made from a combination of the same 20 chemical building blocks, called amino acids. The difference between the proteins that make up bone, blood, hair and eyeballs is largely one of shape.
Genes are the recipes for stringing together amino acids into proteins, but the way in which those strings fold back on themselves determines their shape. So understanding genes’ roles in disease requires understanding how proteins fold.
In a series of recent papers, researchers at MIT’s Computer Science and Artificial Intelligence Laboratory have demonstrated a promising new technique for modeling such protein folding. While not as accurate as some existing techniques, it is much more computationally efficient. Sophisticated, atom-by-atom simulations that run on hundreds of thousands of computers might take months to model a few milliseconds of protein folding. The researchers’ new technique can model the same process in minutes on a single laptop.
Speed is of the essence as the amount of unprocessed genomic data proliferates. “There’s the Broad project, there’s X many species that have been sequenced now, and the sequence data is just vastly outpacing the speed with which you could apply some of these other techniques,” says Charles O’Donnell, a PhD student in the Department of Electrical Engineering and Computer Science who helped develop the new approach. “If you want to make sense of all this high-throughput data that’s coming from this great biotech innovation, then you need something quick.”
Other “quick” methods of simulating protein folding exist, but the MIT researchers’ appears to be more accurate. There is still much we don’t know about the actual structure of proteins, O’Donnell cautions, so that makes assessing the quality of computational methods difficult. But at the 19th Annual International Conference on Intelligent Systems for Molecular Biology (ISMB) in July, the MIT researchers will present a paper demonstrating that for a class of proteins known as amyloids, their technique’s predictions match the currently available data with 81 percent accuracy, whereas high-efficiency techniques previously managed 42 percent at best.
Computational modeling of protein folding has been an active research area for decades, but “it hasn’t been entirely clear whether it was going to be useful or not,” says Susan Lindquist, an MIT professor of biology, recent recipient of the National Medal of Science, and, along with CSAIL’s Bonnie Berger, one of O’Donnell’s faculty advisors. “I think that this paper helps realize that goal.”
Quantity over quality
When a protein folds, amino acids far from each other on the protein strand are brought close together, and chemical bonds form between them. That folding, however, brings other amino acids into proximity with each other, and those acids could exert either an attractive or a repulsive force on each other. Predicting a protein’s shape is a matter of figuring out which regions of the strand could have affinities for each other, and whether bringing those regions together would cause unsupportable tensions elsewhere.
Atom-by-atom simulations can model amino acids’ interactions very precisely, but because they’re so computationally complex, they’ve generally been restricted to protein strands with only a couple dozen amino acids, whereas full proteins can comprise hundreds or even thousands. Making the simulations computationally efficient means sacrificing information about the amino acids’ interactions, and most previous attempts have tried to strike a balance between accuracy of representation and simplicity of description.
MIT computer science professors Bonnie Berger and Srini Devadas, O’Donnell and Jérôme Waldispuhl, a former MIT math instructor who’s now an assistant professor at McGill, adopted a somewhat different approach. They employ what they describe as a “coarse representation” of a protein’s chemical properties, but that allows them to generate a huge number of candidate shapes. Their algorithm then looks for the features that occur most frequently across all the candidates, which it then synthesizes into a small group of likely structures.
Working with collaborators at McGill, Boston College, and the MIT Department of Biology, they’ve applied the technique to several different problems. The paper they’re presenting in July describes their amyloid shape-prediction results, but at the 15th Annual International Conference on Research in Computational Molecular Biology on March 28, they’ll present another paper describing the precise sequence of steps by which different types of proteins — mainly so-called beta-sheet proteins — fold. There, Waldispuhl explains, the trick is that each step in the folding pathway is itself a different shape in the library of candidates, and the algorithm finds a pathway through them. Two years ago at the same conference, the researchers presented an earlier result in which they used their technique to explain the commonalities between proteins with different sequences of amino acids that nonetheless played the same role in certain biological systems, implying that they had structural similarities.
“Protein folding continues to be wide-open problem with desperate need of more rigorous mathematical, statistical and computer-science approaches,” says Sorin Istrail, a professor of computer science at Brown University who specializes in computational biology. What distinguishes the MIT researchers’ work, he says, is its “rigorously mathematical results.” “The world needs to do what Bonnie and Charlie are doing,” Istrail says, “taking one aspect of the problem and building rigorous methods for that particular component.”
This story is republished courtesy of MIT News (), a popular site that covers news about MIT research, innovation and teaching.
Provided by Massachusetts Institute of Technology