September 15, 2017 report
Metal-catalyzed addition of saturated carbon into C-C bonds: A relevant reaction for the synthesis type II polyketides
 Science (2017). DOI: 10.1126/science.aao0453")
(�鶹��Ժ)—Type II polyketides are a class of compounds found in nature as secondary metabolites derived from microorganisms. Their complex structures are characterized by fused polycyclic aromatic rings decorated with a profusion of ketones and hydroxyl groups. Many common antibiotics, anti-fungal, and anti-cancer drugs are type II polyketides or are derivatives of these naturally-occurring compounds. However the "laboratory synthesis" of type II polyketides is often challenging; as in living systems, it is a highly selective multi-step enzymatic process.
Researchers from the University of Texas led by Professor Michael J. Krische have developed a ruthenium-catalyzed cycloaddition of benzocyclobutenones and diols that directly deliver substructures found in diverse type II polyketides. Their catalytic reactions involve successive insertion of saturated diol carbon-hydrogen (C-H) bonds into a carbon-carbon (C-C) bond by way of a ruthenacycle intermediate. Their report appears in Science.
This research builds on prior studies by this group on metal-catalyzed C-C bond forming reactions of alcohols via hydrogen transfer—a modern twist on carbonyl addition chemistry. Whereas classical carbonyl additions typically involve the reaction of pre-formed organometallic reagents with an aldehyde or ketone (e.g., a Grignard reaction), in the current paper these reactive species are generated transiently and in duplicate. The ruthenium catalyst inserts into a stained C-C bond to form a ruthenacycle bearing TWO carbon-ruthenium bonds, and the catalyst converts the diol to a transient diketone. These species engage in double-carbonyl addition that results in cycloaddition. The net result is the formal insertion of two saturated diol C-H bonds into a saturated C-C bond.
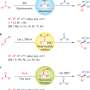
As a model reaction to test their concept, the authors reacted benzocyclobutenone with trans-cyclohexane 1,2-diol in the presence of their ruthenium catalyst. After tweaking the reaction conditions, they obtained an 88% yield of the tricyclic product bearing a bridgehead diol, which formed exclusively as the syn-stereoisomer.
To examine the versatility of their conditions, they reacted a range of benzocyclobutenones with a variety of 1,2-diols. Several of the diols tested had fused rings. This resulted in the formation of cycloadducts with tetracyclic substrates found in several type-II polyketides, including angucycline natural products with congested "bay-region" architectures. Notably, each of the reactions displayed complete regio- and stereoselectivity. Additionally, the reaction tolerated halide substitution on the benzene ring allowing for further functionalization.
Studies to elucidate the reaction mechanism showed that the cycloaddition can be performed in a redox-independent manner. That is, the benzocyclobutenone can react with a diol, a ketol, or a dione producing the same product as a single regio- and stereoisomer. While the reaction of the diol is oxidative and requires a slight excess of benzocyclobutenone, the reaction of the ketol is redox-neutral and can be performed with equal amounts of benzocyclobutenone and ketol. The reductive reaction of the dione requires the addition of 2-propanol to act as a reductant
Beyond broadening access to type II polyketides, the reactivity embodied by these studies has broader implications in the field of chemical synthesis.
Dr. Michael Krische told �鶹��ԺOrg, "In contrast to progress recently made in the field of C-H activation, the development of catalysts that modify C-C sigma-bonds has lagged behind, and is largely restricted to intramolecular reactions that insert tethered π-bonds, often requiring directing-groups. Hydrogen-transfer offers a broad, new approach to the functionalization of C-C bonds using abundant, renewable alcohols as coupling partners."
More information: Matthias Bender et al. Ruthenium-catalyzed insertion of adjacent diol carbon atoms into C-C bonds: Entry to type II polyketides, Science (2017).
Abstract
Current catalytic processes involving carbon-carbon bond activation rely on π-unsaturated coupling partners. Exploiting the concept of transfer hydrogenative coupling, we report a ruthenium(0)-catalyzed cycloaddition of benzocyclobutenones that functionalizes two adjacent saturated diol carbon-hydrogen bonds. These regio- and diastereoselective processes enable convergent construction of type II polyketide substructures.
Journal information: Science
© 2017 �鶹��Ժ