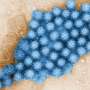
Fig. 1. KMAP visualization of SELEX-seq data for the transcription factor MAFK. Each dot represents a k-mer. Clusters of k-mers at the center correspond to DNA motifs, while the surrounding dots represent random k-mers. Each central cluster represents a distinct motif, with red dots highlighting k-mers from the major motif. Credit: Genome Research (2025). DOI: 10.1101/gr.279458.124
Researchers from the University of Eastern Finland, Aalto University and the University of Oulu have developed a new computational method for exploring DNA sequence patterns. The method, called KMAP, enables intuitive visualization of short DNA sequences and helps reveal how regulatory elements behave in different biological contexts. The study was recently in Genome Research.
KMAP projects DNA sequences—known as k-mers—into two-dimensional space, making it easier to identify and interpret biologically significant DNA sequence patterns, also called DNA motifs (fig. 1). In a re-analysis of Ewing sarcoma data, the researchers used KMAP to analyze genomic regions involved in gene regulation.
Fig. 2. Schematic illustration of enhancer regulation in Ewing sarcoma versus the healthy state. In Ewing sarcoma (top), the transcriptional repressor ETV6 competitively binds to transcription factor FLI1 binding sites and closes the enhancer regions, contributing to disease progression. Upon ETV6 degradation (bottom), the enhancer becomes accessible in the presence of FLI1 alone, allowing other transcription factors—BACH1, OTX2, KCNH2, and a potential unknown TF—to bind. These factors often co-localize within a window of about 70 base pairs near the motif CCCAGGCTGGAGTGC and may function jointly in regulating gene expression. Credit: Genome Research (2025). DOI: 10.1101/gr.279458.124
They found that the transcription factors BACH1, OTX2 and KCNH2/ERG1 were suppressed by the oncogene ETV6 and became active at promoter and enhancer regions once ETV6 was degraded (fig. 2). Notably, the study also identified an uncharacterized DNA motif, CCCAGGCTGGAGTGC, which frequently co-localized with BACH1 and OTX2 within a short window in enhancer regions. This spatial clustering suggests a potential new regulatory element relevant to cancer biology.
KMAP was also used to analyze the outcomes of a genome editing experiment, where the widely used CRISPR-Cas9 technique was applied to a specific location in the human genome called the AAVS1 locus. After editing, cells naturally repair the broken DNA in different ways.
By visualizing thousands of DNA sequences from this process, KMAP revealed four common patterns of how the DNA was repaired—each associated with a distinct repair pathway used by the cell. Understanding these patterns can help researchers design more precise gene-editing strategies and predict the types of edits that are most likely to occur.
"KMAP offers a more intuitive way to investigate motifs in DNA sequence data," says the study's lead author, Dr. Lu Cheng from the University of Eastern Finland. "By visualizing the distribution of short DNA sequences, we can better interpret regulatory patterns and understand how they change in different biological conditions."
"KMAP is a versatile tool that can be applied to many types of sequencing data," says Professor Gonghong Wei from the University of Oulu. "In cancer research, it can help identify regulatory elements from ChIP-seq data, and it also holds promise for studying RNA-binding proteins and their binding preferences. Its ability to reveal structure in complex sequence data makes it broadly useful across molecular biology."
This collaborative work demonstrates how computational biology can uncover hidden layers of gene regulation and support future research in cancer and genome engineering.
More information: Chengbo Fu et al, k-mer manifold approximation and projection for visualizing DNA sequences, Genome Research (2025).
Journal information: Genome Research
Provided by University of Eastern Finland