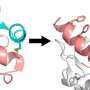
Graphic depicts the predicted multimer structure (via AlphaFold) of the enzyme in complex with one example of peptide substrates. PapB is a complex metalloenzyme that uses cysteine residues to coordinate to Iron-Sulfur complexes. Credit: Sethera Therapeutics
A team of researchers from the University of Utah and Sethera Therapeutics has uncovered a powerful new way to build more stable and drug-like peptides, opening the door to medicines that could target diseases long considered "undruggable." Their findings are last week in the Proceedings of the National Academy of Sciences.
In the paper titled, "Diverse thioether macrocyclized peptides through a radical SAM maturase," the team studied a natural enzyme, called PapB, that can "staple" peptides into circular structures known as macrocycles. What makes PapB so unusual is that it combines flexibility and precision: It works on many different building blocks—including those that biology usually rejects—yet still creates a single, predictable bond. In one gentle step, it transforms linear peptides into sturdy, ring-shaped molecules that are more stable, resistant to breakdown, and better suited for drug development.
Many peptide drugs are stabilized with disulfide bonds, which break down in the body, or rely on complicated, costly, and time-consuming chemical methods to achieve the same effect. The PapB, however, streamlines the process, creating durable "stapled" peptides that drug developers can program with unprecedented ease. This opens vast new chemical space for peptide medicines, including scaffolds associated with better cell penetration and oral dosing—two qualities essential for advancing peptide therapeutics.
PapB overcomes these issues by combining breadth with precision, delivering a programmable, one-step solution for generating robust peptide macrocycles that broaden the landscape for therapeutic design.
According to the lead author, Karsten Eastman, CEO and Co-founder of Sethera Therapeutics, "Peptides that behave both like small molecules and biologics at the same time—that's the goal. This enzyme lets us program a durable thioether 'staple' across an unusually wide range of backbones in a single enzymatic step, massively expanding the design space we can test against difficult-to-hit biological targets.
"For discovery teams, that means faster iteration, richer and more diverse libraries, and scaffolds with the stability and permeability profiles needed to move from an intriguing hit to a viable therapeutic lead."
A new horizon for peptide therapeutics
This breakthrough positions PapB as a sequence-agnostic thioether ligase, opening unprecedented chemical space for peptide drug discovery. By bridging the gap between biological selectivity and chemical flexibility, Sethera and University of Utah researchers are enabling next-generation peptide therapeutics aimed at targets previously considered "undruggable."
"What's unusual here is not just promiscuity… it is promiscuity with control. PapB accepts D- and β-amino acids and even N-methylated backbones, yet still places a single thioether exactly where the chemistry demands. That combination opens a practical route to stable, macrocyclic peptide scaffolds that were previously difficult to impossible to access with synthetic methods alone," said Vahe Bandarian, Ph.D., Professor of Chemistry, University of Utah; CSO and Co-Founder, Sethera Therapeutics.
With this discovery, researchers now have a programmable, one-step method for making peptide macrocycles that combine stability, diversity, and drug-like properties. The breakthrough offers a powerful new tool for biotech and pharma teams seeking next-generation treatments in areas where traditional drugs have failed.
More information: Karsten A. S. Eastman et al, Diverse thioether macrocyclized peptides through a radical SAM maturase, Proceedings of the National Academy of Sciences (2025).
Journal information: Proceedings of the National Academy of Sciences
Provided by Sethera Therapeutics